Η SMA προκαλείται από την έλλειψη του λειτουργικού γονιδίου SMN1, που έχει ως αποτέλεσμα τα βρέφη που πάσχουν από την ασθένεια να μην παράγουν μια βασική πρωτεΐνη που είναι απαραίτητη για την επιβίωση των κινητικών νευρώνων τους.
Ως συνέπεια, αμέσως μετά τη γέννηση, προκαλείται προοδευτικά μη αναστρέψιμη απώλεια των κινητικών νευρώνων που επηρεάζουν όλες τις μυϊκές λειτουργίες, συμπεριλαμβανομένης της αναπνοής, της κατάποσης και της βασικής κίνησης.
Η συχνότητα εμφάνισης της SMA είναι περίπου 1 στις 10.000 γεννήσεις παγκοσμίως και περίπου 1 στα 54 άτομα φέρουν το γενετικό ελάττωμα. Κάθε χρόνο, υπολογίζεται ότι στην Ευρώπη γεννιούνται περίπου 550-600 βρέφη με SMA και περίπου 8 βρέφη στην Ελλάδα.
Η φροντίδα και η θεραπεία παιδιών με SMA είναι μια σημαντική επιβάρυνση για το σύστημα υγείας στην Ευρώπη, με το σωρευτικό εκτιμώμενο κόστος της υγειονομικής περίθαλψης ανά παιδί να κυμαίνεται μεταξύ 2,5 και 4 εκατομμυρίων ευρώ τα πρώτα 10 κιόλας χρόνια.
Τύποι της SMA και επιστημονικές εξελίξεις
Υπάρχουν τρεις βασικοί τύποι της SMA:
• Η SMA Τύπου 1 είναι η πιο σοβαρή μορφή της ασθένειας και αντιπροσωπεύει το 60% των περιπτώσεων. Τα παιδιά με SMA Τύπου 1, εάν δεν υποβληθούν σε θεραπεία, χάνουν την ικανότητα κίνησης μόλις τους πρώτους 6 μήνες ζωής, ενώ περισσότερο από το 90% των περιπτώσεων καταλήγουν ή χρειάζονται μόνιμη αναπνευστική υποστήριξη πριν από το δεύτερο έτος της ηλικίας.
• Η SMA Τύπου 2 αντιπροσωπεύει το 30% των περιπτώσεων. Τα παιδιά με SMA Τύπου 2 δε θα μπορέσουν ποτέ να στέκονται χωρίς υποστήριξη και συχνά χρειάζονται αναπηρικό καροτσάκι. Μπορεί να είναι σε θέση να καθίσουν χωρίς βοήθεια στην πρώιμη φάση της ανάπτυξής τους, αλλά συχνά χάνουν αυτή την ικανότητα από τα μέσα της εφηβείας.
• Η SMA Τύπου 3 αντιπροσωπεύει το 10% των περιπτώσεων. Τα άτομα με SMA Τύπου 3 δυσκολεύονται να περπατήσουν, να τρέξουν και να ανεβοκατεβαίνουν σκάλες. Επιπλέον, μπορεί να χάσουν την ικανότητα να στέκονται ή να περπατούν χωρίς υποστήριξη με την πάροδο του χρόνου.
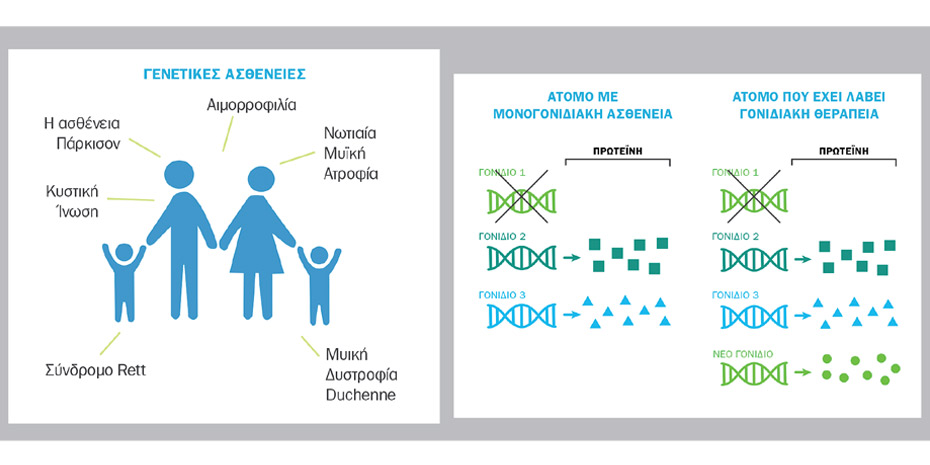
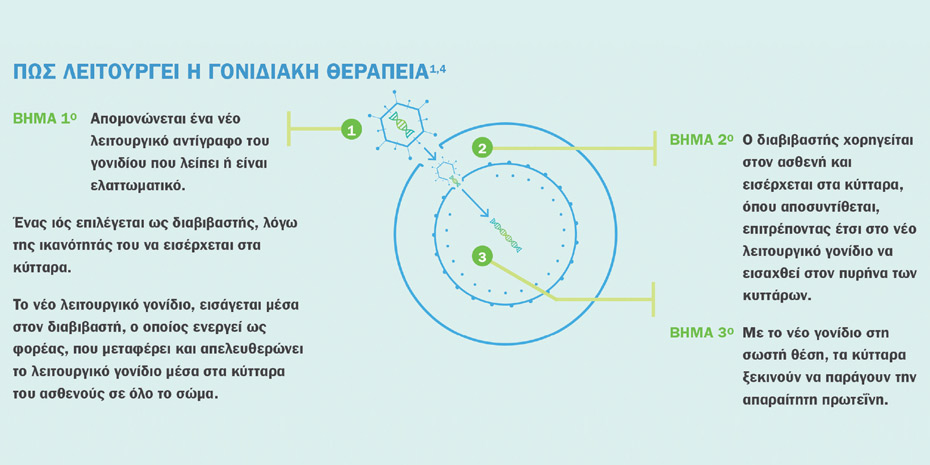
Σήμερα υπάρχουν δύο εγκεκριμένες θεραπευτικές επιλογές. H μία στηρίζεται σε φαρμακευτική αγωγή που χορηγείται σε επαναλαμβανόμενες δόσεις συντήρησης σε βάθος χρόνου, για το υπόλοιπο της ζωής του ασθενούς. Η δεύτερη είναι μια εφάπαξ γονιδιακή θεραπεία, η οποία σχεδιάστηκε για να αντιμετωπίσει τη γενετική αιτία της νόσου, αντικαθιστώντας στα κύτταρα του ασθενούς τη λειτουργία του γονιδίου SMN1, που λείπει ή δεν λειτουργεί, με ένα νέο λειτουργικό αντίγραφο, αναστέλλοντας την πρόοδο της νόσου. Κυκλοφορεί εδώ και ένα χρόνο στην Αμερική και εγκρίθηκε τον Μάιο 2020 και για τις χώρες της Ευρωπαϊκής Ένωσης.
Νεογεννητικός έλεγχος για SMA
Η έγκαιρη διάγνωση της νόσου δεν είναι εύκολη, καθώς τα πρώτα σημάδια της SMA είναι ανεπαίσθητα, με τα παιδιά να φαίνονται υγιή ενόσω καταστρέφονται οι κινητικοί νευρώνες. Για τον σκοπό αυτό, κρίνεται απαραίτητη η ανάπτυξη ενός γενικευμένου προγράμματος ελέγχου για SMA σε όλες τις κυήσεις, πριν ή μετά τον τοκετό.
Έτσι, διασφαλίζεται η έγκαιρη ανίχνευση της SMA, ώστε να χορηγείται άμεσα η θεραπεία και να αποφεύγεται κατά το μέγιστο δυνατό η καταστροφή των κινητικών νευρώνων στα νεογνά που νοσούν.
Σημειώνεται πως σήμερα έχει εγκριθεί η υπαγωγή όλων των σπάνιων ασθενειών για τις οποίες υπάρχει θεραπεία, όπως η SMA, σε έλεγχο νεογνών.
Η αξία της γονιδιακής θεραπείας
Η φροντίδα και θεραπεία παιδιών με SMA αποτελεί σημαντική επιβάρυνση για το σύστημα υγείας. Επιπλέον, υπάρχουν σημαντικές επιπτώσεις στα μέλη της οικογένειας, οι οποίες δεν μπορούν να ποσοτικοποιηθούν, όπως οι αυξημένες ανάγκες για φροντίδα, οι περιορισμοί στις επιλογές σταδιοδρομίας και εξέλιξης, το κοινωνικό και συναισθηματικό βάρος.
Η νέα γονιδιακή θεραπεία χορηγείται εφάπαξ και διαφοροποιείται από το τρέχον θεραπευτικό πρότυπο, το οποίο βασίζεται σε χρόνια θεραπεία, για όλη τη διάρκεια της ζωής ενός ασθενούς.
ΣΗΜΕΙΩΣΗ: Αυτές οι πληροφορίες προορίζονται για γενική πληροφόρηση και ενημέρωση του κοινού και σε καμία περίπτωση δεν μπορούν να υποκαταστήσουν τη συμβουλή ιατρού ή άλλου αρμόδιου επαγγελματία υγείας